DownloadAssemblies - Download assemblies and their metadata from specified databases
Description
The DownloadAssemblies module facilitates the download of genome assemblies. It parses command-line arguments to initiate the download process, allowing users to efficiently retrieve assemblies from either the NCBI or ENA661k databases. The downloaded assemblies are stored in the specified output directory. It supports filtering based on user-defined criteria and the retrieval of associated metadata.
Features
Parallel downloading of assemblies using multiple threads.
Support for downloading genome assemblies from the NCBI and ENA661k databases.
Filtering of assemblies based on criteria such as genome size, contig number, assembly level, etc.
Sample metadata retrieval.
Option to download assemblies based on taxon name or a table with sample IDs.
Dependencies
NCBI datasets command-line too installed (instructions here).
Usage
The DownloadAssemblies module can be used as follows:
SR DownloadAssemblies -f /path/to/input_file_with_taxon -db NCBI ENA661K -o /path/to/output -e email@example -th 4 -fm --download
Command-Line Arguments
-db, --database
(Required) Databases from which assemblies will be downloaded.
Choices: NCBI, ENA661K
-o, --output-directory
(Required) Path to the output directory.
-e, --email
(Required) Email provided to Entrez.
-f, --input-file
(Required) TSV file containing filtering parameters and either a path to a list of accession numbers (only fo NCBI) or the taxon applied before assembly download.
-th, --threads
(Optional) Number of threads used for download. You should provide an API key to perform more requests through Entrez.
Default: 1
-r, --retry
(Optional) Maximum number of retries when a download or request fails.
Default: 7
-k, --api-key
(Optional) Personal API key provided to the NCBI. If not set, only 3 requests per second are allowed through Entrez. With a valid API key the limit increases to 10 requests per second.
-fm, --fetch-metadata
(Optional) If provided, the process downloads metadata for the assemblies.
Default: False
--download
(Optional) If the assemblies that passed the filtering criteria should be downloaded.
--nocleanup
(Optional) Flag to indicate whether to skip cleanup after running the module.
--debug
(Optional) Flag to indicate whether to run the module in debug mode.
Default: False
--logger
(Optional) Path to the logger file.
Default: None
Algorithm Explanation
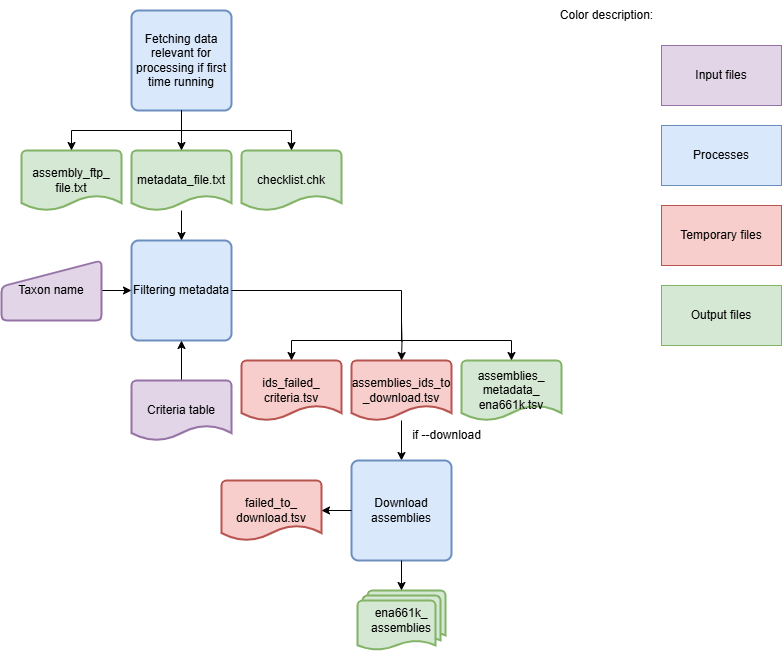
The DownloadAssemblies Workflow is shown in the flowchart below:
Workflow for downloading assemblies from NCBI

Workflow for downloading assemblies from ENA661K
Workflow for downloading metadata
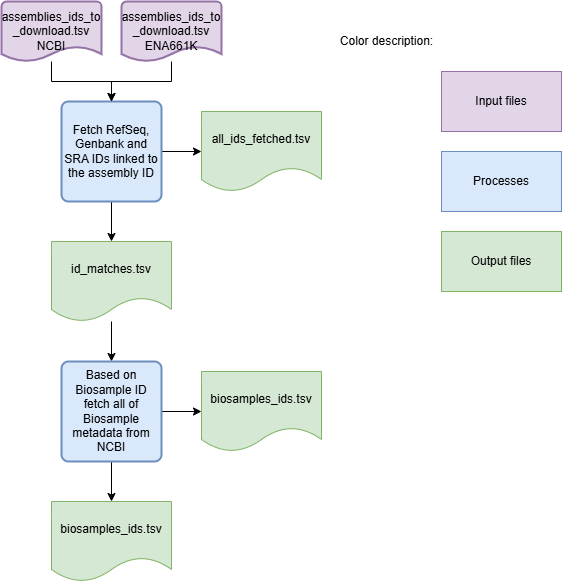
To download genome assemblies from both databases, the first step is to download the sample metadata. For the NCBI database, the sample metadata is downloaded and filtered according to the selection criteria specified by the user. For the ENA661k database, a metadata table for all samples is available. Schema Refinery identifies the lines matching the samples and filters them based on the user-defined criteria. After the metadata is filtered, the assemblies are downloaded. If the –nocleanup` argument is not used, the final output is the log file and a ZIP archive containing the genome assemblies. Use the option –fetch-metadata to get an output file with more complete metadata.
Filtering criteria example
The filtering criteria are provided through a TSV file with the following columns:
taxon: Scientific name of the taxon (NCBI, ENA661K)
input_table: Text file with a list of accession numbers for the NCBI Assembly database (NCBI)
abundance: Abundance of the taxon in the environment (ENA661K) (0-1)
genome_size: Genome size in Mbp (NCBI, ENA661K) (>1)
size_threshold: Threshold for genome size in Mbp (NCBI, ENA661K) (0-1)
max_contig_number: Maximum number of contigs in the assembly (NCBI, ENA661K) (>1)
known_st: Known sequence type (ENA661K) (True, False, None)
any_quality: Any quality (ENA661K) (True, False, None)
ST_list_path: Path to the sequence type list (ENA661K) (path, None)
assembly_level: Assembly level (NCBI, ENA661K) (chromosome,complete,contig,scaffold)
reference: If reference genome (NCBI) (True, False, None)
assembly_source: Assembly source (NCBI) (all, RefSeq, GenBank)
file_to_include: files to include (NCBI) (genome, rna, protein, cds, gff3, gtf, gbff, seq-report, none)
verify_status: Verify status (NCBI) (True, False, None)
exclude_atypical: Exclude atypical (NCBI) (True, False, None)
Some filtering criteria file are specific to one of the databases (e.g. ST_list_path is only valid for the ENA661K database). When None or an empty value is provided, the filtering criteria will not be applied. The file can only contain either the taxon or input_table option, not both. An example of this file exists in the DownloadAssemblies folder here. Use it as a template to create your own criteria files.
Outputs
The folder and file structure of the output directory is shown below.
OutputFolderName
├── assemblies_ncbi.zip # -db NCBI --download
├── ena661k_assemblies # -db ENA661 --download
│ ├── x.contigs.fa.gz
│ ├── y.contigs.fa.gz
│ ├── z.contigs.fa.gz
│ └── ...
├── metadata_all # -fm
│ ├── biosamples_ids.tsv
│ ├── id_matches.tsv
│ ├── all_ids_fetched.tsv
│ └── metadata_biosamples.tsv
├── assemblies_metadata_ena661k.tsv # -db ENA661k
├── assemblies_metadata_ncbi.tsv # -db NCBI
├── metadata_ncbi # -db NCBI --nocleanup
│ ├── assemblies_ids_to_download.tsv
│ └── ids_failed_criteria.tsv
└── metadata_ena661k # -db ENA661k --nocleanup
├── assemblies_ids_to_download.tsv
├── failed_to_download.tsv
└── id_failed_criteria.tsv
Examples
Here are some example commands to use the DownloadAssemblies module:
# Download assemblies from NCBI for a specific taxon
SR DownloadAssemblies -f /path/to/input_file_with_taxon -db NCBI -o /path/to/output -e email@example.com -th 4 --download
# Download assemblies from NCBI using an IDs table
SR DownloadAssemblies -f /path/to/input_file_with_id_table -db NCBI -o /path/to/output -e email@example.com -th 4 --download
# Download assemblies from both NCBI and ENA661K with filtering criteria
SR DownloadAssemblies -t "Streptococcus pyogenes" -db NCBI ENA661K -o /path/to/output -e email@example.com -th 4 -fm --download
Troubleshooting
If you encounter issues while using the DownloadAssemblies module, consider the following troubleshooting tips:
Ensure that you have a stable internet connection.
Verify that your email and API key (if provided) are correct.
Check the output directory for any error logs or messages.
Check if the versions of the dependencies within the environment are compatible.
Increase the number of retries using the -r or –retry option if downloads are failing.